deepumaj1 wrote:Hi. Can you give a little more details about your simulation set up? Also, if possible, can you give a snapshot of the thickening borders? Do you see any spreading of the interface??
Deepu
Dear Depeu
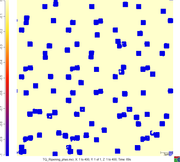
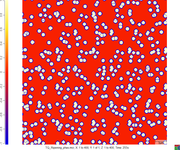
I want to get the growing-up like image1 rather than image2.and the setting is listed as follow
# Time input data
# ===============
# Finish input of output times (in seconds) with 'end_of_simulation'
# 'regularly-spaced' outputs can be set with 'linear_step'
# or 'logarithmic_step' and then specifying the increment
# and end value
# ('automatic_outputs' optionally followed by the number
# of outputs can be used in conjuction with 'linear_from_file')
# 'first' : additional output for first time-step
# 'end_at_temperature' : additional output and end of simulation
# at given temperature
0.001
linear_step 0.01 2
end_of_simulation
# Time-step?
# Options: fix ...[s] automatic automatic_limited
automatic
# Coefficient for phase-field criterion 1.00
# Coefficient for segregation criterion 0.900
# Number of steps to adjust profiles of initially sharp interfaces [exclude_inactive]?
20
#
# Phase data
# ==========
# Number of distinct solid phases?
1
#
# Data for phase 1:
# -----------------
# Simulation of recrystallisation in phase 1?
# Options: recrystall no_recrystall [verbose|no_verbose]
no_recrystall
# Is phase 1 anisotrop?
# Options: isotropic anisotropic faceted antifaceted
isotropic
# Should grains of phase 1 be reduced to categories?
# Options: categorize no_categorize
categorize
#
# Orientation
# -----------
#
# Grain input
# ===========
# Type of grain positioning?
# Options: deterministic random [deterministic_infile] from_file
deterministic
# NB: the origin of coordinate system is the bottom left-hand corner,
# all points within the simulation domain having positive coordinates.
# Number of grains at the beginning?
4
# Input for grain type 1
# ----------------------
# Geometry of grain type 1
# Options: round rectangular elliptic round_inverse
round
# Center x,z coordinates [micrometers], grain number 1?
10
10
# Grain radius? [micrometers]
0.3
# Shall grain type 1 be stabilized or shall
# an analytical curvature description be applied?
# Options: stabilisation analytical_curvature
stabilisation
# Should the Voronoi criterion for grains of type 1 be applied?
# Options: voronoi no_voronoi
no_voronoi
# Phase number? (int)
1
# Input data for grain number 2:
# Geometry?
# Options: round rectangular elliptic round_inverse
round
# Center x,z coordinates [micrometers], grain number 2?
30
10
# Grain radius? [micrometers]
0.3
# Shall grain type 1 be stabilized or shall
# an analytical curvature description be applied?
# Options: stabilisation analytical_curvature
stabilisation
# Should the Voronoi criterion for grains of type 1 be applied?
# Options: voronoi no_voronoi
no_voronoi
# Phase number? (int)
1
# Input for grain type 3
# ----------------------
# Geometry of grain type 3
# Options: round rectangular elliptic round_inverse
round
# Center x,z coordinates [micrometers], grain number 3?
10
30
# Grain radius? [micrometers]
0.3
# Shall grain type 3 be stabilized or shall
# an analytical curvature description be applied?
# Options: stabilisation analytical_curvature
stabilisation
# Should the Voronoi criterion for grains of type 3 be applied?
# Options: voronoi no_voronoi
no_voronoi
# Phase number? (int)
1
# Input for grain type 4
# ----------------------
# Geometry of grain type 4
# Options: round rectangular elliptic round_inverse
round
# Center x,z coordinates [micrometers], grain number 4?
30
30
# Grain radius? [micrometers]
0.3
# Shall grain type 4 be stabilized or shall
# an analytical curvature description be applied?
# Options: stabilisation analytical_curvature
stabilisation
# Should the Voronoi criterion for grains of type 4 be applied?
# Options: voronoi no_voronoi
voronoi
# Phase number? (int)
1
#
#
#
# Data for further nucleation
# ===========================
# Enable further nucleation?
# Options: nucleation nucleation_symm no_nucleation [verbose|no_verbose]
no_nucleation
#
# Phase interaction data
# ======================
#
# Data for phase interaction 0 / 1:
# ---------------------------------
# Simulation of interaction between phase 0 and 1?
# Options: phase_interaction no_phase_interaction
# [standard|particle_pinning[_temperature]|solute_drag]
# | [redistribution_control]
phase_interaction
# 'DeltaG' options: default
# avg ... [] max ... [J/cm**3] smooth ... [degrees] noise ... [J/cm**3]
avg 1 max 20 smooth 15
# I.e.: avg +1.00 smooth +15.0 max +2.00000E+01
# Type of interfacial energy definition between phases 0 and 1?
# Options: constant temp_dependent
constant
# Interfacial energy between phases phases 0 and 1? [J/cm**2]
# [max. value for num. interface stabilisation [J/cm**2]]
5E-6
# Type of mobility definition between phases phases 0 and 1?
# Options: constant temp_dependent dg_dependent [fixed_minimum]
constant
# Kinetic coefficient mu between phases phases 0 and 1 [ min. value ] [cm**4/(Js)] ?
4e-6
# Data for phase interaction 1 / 1:
# ---------------------------------
# Simulation of interaction between phase 1 and 1?
# Options: phase_interaction no_phase_interaction identical phases nb
# [standard|particle_pinning[_temperature]|solute_drag]
# | [redistribution_control]
phase_interaction
# Type of interfacial energy definition between phases 1 and 1?
# Options: constant temp_dependent
constant
# Interfacial energy between phases phases 1 and 1? [J/cm**2]
# [max. value for num. interface stabilisation [J/cm**2]]
5E-4
# Type of mobility definition between phases phases 1 and 1?
# Options: constant temp_dependent dg_dependent [fixed_minimum]
constant
# Kinetic coefficient mu between phases phases 1 and 1 [ min. value ] [cm**4/(Js)] ?
2.96e-10
#
#
#
# Concentration data
# ==================
# Number of dissolved constituents? (int)
1
# Type of concentration?
# Options: atom_percent (at%)
# weight_percent (wt%)
atom_percent
#
#
# Diffusion Data
# --------------
# ["Terse Mode": Each line starts with component number and phase number]
# Options: diagonal [x] multi [y(1..k)]
# x: one of the characters "n", "d", "g", "l", "z", "i", "I", or "f"
# y: chain of "n", "d", "g", "l", "z", or "f" (for each component)
# default: "g" resp. "gggg..."
# Rem: "n":no diffusion, "d": input, "f": T-dep. from file
# "i":infinite, "I": infinite in each grain
# from database: "g": global, "l": local, "z" global z-segmented
# Extra option [+b] for grain-boundary diffusion
# Extra line option (prefactor on time step): cushion <0-1>
# Extra line option: infinite_limit [cm**2/s]
# Extra line option: maxfactor_local [real > 1.0] (default: 10.0)
# Extra line option: factor [real > 0.]
# Finish input of diffusion data with 'end_diffusion_data'.
#
# How shall diffusion of component 1 in phase 0 be solved?
diff
# Diff.-coefficient:
# Prefactor? (real) [cm**2/s]
5.00000E-06
# Activation energy? (real) [J/mol]
0.0000
# How shall diffusion of component 1 in phase 1 be solved?
diff
# Diff.-coefficient:
# Prefactor? (real) [cm**2/s]
1.00000E-12
# Activation energy? (real) [J/mol]
0.0000
#
#
#
# Phase diagram - input data
# ==========================
#
# List of phases and components which are stoichiometric:
# phase and component(s) numbers
# List of concentration limits (at%):
# <Limits>, phase number and component number
# List for ternary extrapolation (2 elements + main comp.):
# <interaction>, component 1, component 2
# Switches: <stoich_enhanced_{on|off}> <solubility_{on|off}>
# End with 'no_more_stoichio' or 'no_stoichio'
no_stoichio